核心摘要
- 美国 FDA 已授予 Kerendia 针对伴有慢性肾脏病的 1 型糖尿病患者补充申请的优先审评资格。
- 该申请基于 III 期临床数据,结果显示,与安慰剂相比,治疗六个月后患者的尿白蛋白与肌酐比值降低了 25%。
- 若获批准,Kerendia 将成为该特定患者群体(约占美国 1 型糖尿病成人的 20% 至 30%)中首个此类药物。
核心摘要
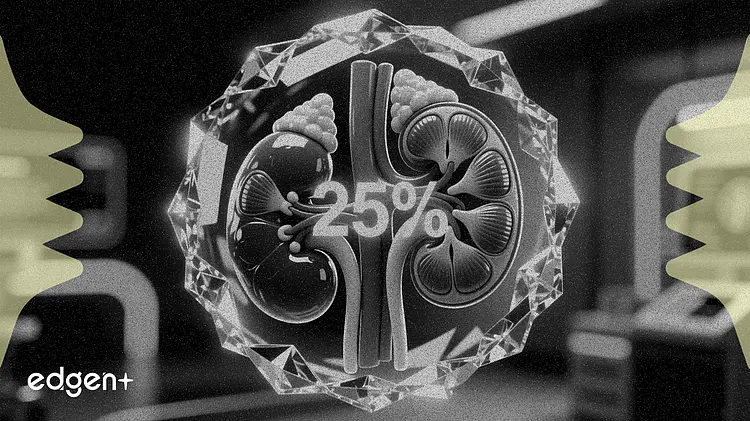
拜耳公司(Bayer AG)周四获得美国食品药品监督管理局(FDA)对其药物 Kerendia 的优先审评认定,旨在将其用途扩展到患有慢性肾脏病和 1 型糖尿病的成年患者。此前的一项试验显示,该药能带来 25% 的肾脏获益。
拜耳执行医学总监 Carolina Aldworth 表示:“FDA 对该申请的受理,凸显了我们正在进行的 KERENDIA 计划及其不断增长的证据库在心血管和肾脏疾病广泛患者群体中的临床重要性。”
该补充新药申请(sNDA)得到了包含 242 名患者的 FINE-ONE III 期研究的支持。该试验达到了主要终点,结果显示,与安慰剂相比,非奈利酮在六个月内将尿白蛋白与肌酐比值(UACR)的最小二乘均值降低了 25%(p<0.001),而 UACR 是衡量肾脏疾病进展的关键指标。
优先审评缩短了 FDA 的评估时间表,有望加速首个盐皮质激素受体拮抗剂(MRA)面向约占美国 1 型糖尿病患者 20% 至 30% 且患有慢性肾脏病的人群上市。这项成功的试验是自 20 世纪 90 年代以来首个针对这一特定患者群体的试验。
FINE-ONE 试验的安全概况与 Kerendia 在 2 型糖尿病患者中已知的概况基本一致。与安慰剂(3.3%)相比,非奈利酮组观察到高钾血症(钾水平升高)的频率更高(10.1%),导致该药的治疗中断率为 1.7%。
Kerendia(通用名为非奈利酮)已经是拜耳的一款核心药物。它于 2021 年首次获批用于治疗与 2 型糖尿病相关的慢性肾脏病,随后于 2025 年获批用于治疗某些类型的心力衰竭,与阿斯利康的 Farxiga 等药物展开竞争。
如果获得批准,标签扩展将开辟一条急需的新治疗途径,并为拜耳的关键医药资产之一带来新的收入来源。投资者现在将关注 FDA 的最终决定,预计将在为期六个月的优先审评窗口内做出。
本文仅供参考,不构成投资建议。