核心摘要
- 美國 FDA 已授予 Kerendia 針對伴有慢性腎臟病的 1 型糖尿病患者補充申請的優先審查資格。
- 該申請基於 III 期臨床數據,結果顯示,與安慰劑相比,治療六個月後患者的尿白蛋白與肌酐比值降低了 25%。
- 若獲批准,Kerendia 將成為該特定患者群體(約佔美國 1 型糖尿病成人的 20% 至 30%)中首個此類藥物。
核心摘要
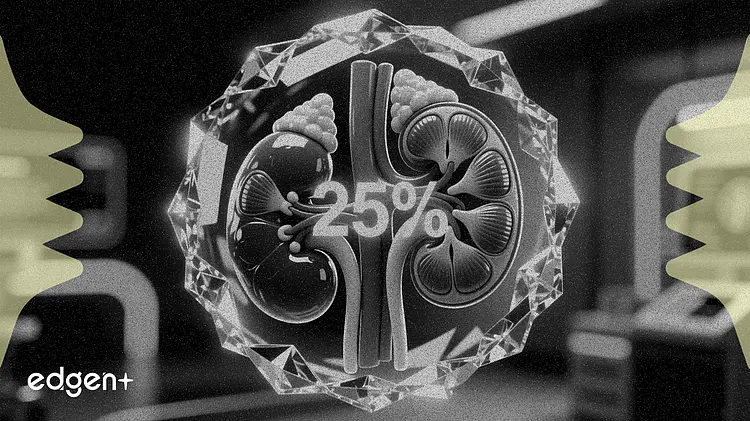
拜耳公司(Bayer AG)週四獲得美國食品藥物管理局(FDA)對其藥物 Kerendia 的優先審查認定,旨在將其用途擴展到患有慢性腎臟病和 1 型糖尿病的成年患者。此前的一項試驗顯示,該藥能帶來 25% 的腎臟獲益。
拜耳執行醫學總監 Carolina Aldworth 表示:「FDA 對該申請的受理,凸顯了我們正在進行的 KERENDIA 計劃及其不斷增長的證據庫在心血管和腎臟疾病廣泛患者群體中的臨床重要性。」
該補充新藥申請(sNDA)得到了包含 242 名患者的 FINE-ONE III 期研究的支持。該試驗達到了主要終點,結果顯示,與安慰劑相比,非奈利酮在六個月內將尿白蛋白與肌酐比值(UACR)的最小二乘均值降低了 25%(p<0.001),而 UACR 是衡量腎臟疾病進展的關鍵指標。
優先審查縮短了 FDA 的評估時間表,有望加速首個鹽皮質激素受體拮抗劑(MRA)面向約佔美國 1 型糖尿病患者 20% 至 30% 且患有慢性腎臟病的人群上市。這項成功的試驗是自 20 世紀 90 年代以來首個針對這一特定患者群體的試驗。
FINE-ONE 試驗的安全概況與 Kerendia 在 2 型糖尿病患者中已知的概況基本一致。與安慰劑(3.3%)相比,非奈利酮組觀察到高鉀血症(鉀水平升高)的頻率更高(10.1%),導致該藥的治療中斷率為 1.7%。
Kerendia(通用名為非奈利酮)已經是拜耳的一款核心藥物。它於 2021 年首次獲批用於治療與 2 型糖尿病相關的慢性腎臟病,隨後於 2025 年獲批用於治療某些類型的心力衰竭,與阿斯特捷利康的 Farxiga 等藥物展開競爭。
如果獲得批准,標籤擴展將開闢一條急需的新治療途徑,並為拜耳的關鍵醫藥資產之一帶來新的收入來源。投資者現在將關注 FDA 的最終決定,預計將在為期六個月的優先審查窗口內做出。
本文僅供參考,不構成投資建議。