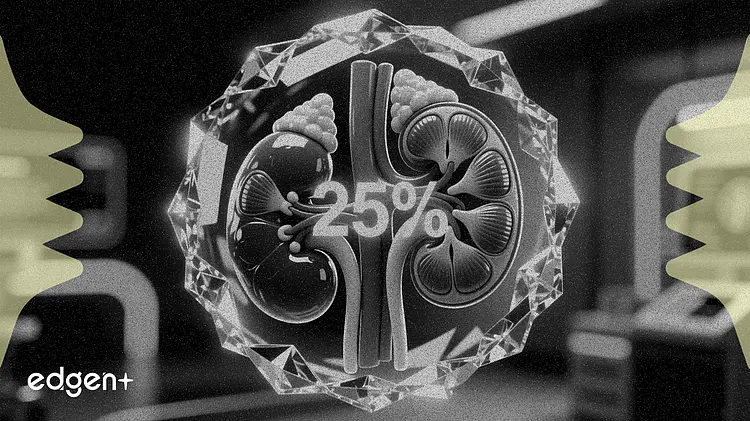바이엘(Bayer AG)은 목요일 미국 식품의약국(FDA)으로부터 자사 약물인 케렌디아(Kerendia)에 대해 우선 심사(Priority Review) 지정을 받았다고 발표했습니다. 이번 승인은 신장 기능에 25%의 개선 효과를 보인 임상 시험 결과를 바탕으로, 만성 신장병을 동반한 성인 1형 당뇨병 환자로 사용 범위를 확대하는 것을 목표로 합니다.
바이엘의 캐롤라이나 알드워스(Carolina Aldworth) 총괄 의학 이사는 "FDA의 이번 신청 접수는 심혈관 및 신장 질환의 광범위한 환자군에 걸쳐 진행 중인 케렌디아 프로그램의 임상적 중요성과 입증된 근거가 강화되고 있음을 강조하는 것"이라고 말했습니다.
이번 보충적 신약 승인 신청(sNDA)은 242명의 환자를 대상으로 한 FINE-ONE 임상 3상 연구를 근거로 합니다. 해당 임상은 신장 질환 진행의 핵심 지표인 소변 알부민-크레아티닌 비(UACR)를 위약군 대비 6개월 동안 최소제곱평균 25% 감소시켜 1차 평가지표를 충족했습니다(p<0.001).
우선 심사는 FDA의 평가 기간을 단축시켜, 만성 신장병을 앓고 있는 미국 내 1형 당뇨병 환자의 20~30%에게 최초의 무기질코르티코이드 수용체 길항제(MRA)를 신속하게 보급할 수 있게 합니다. 이번 임상 성공은 1990년대 이후 이 특정 환자군을 대상으로 한 최초의 사례입니다.
FINE-ONE 임상에서의 안전성 프로파일은 2형 당뇨병 환자에게서 확인된 케렌디아의 기존 프로파일과 대체로 일치했습니다. 혈중 칼륨 수치가 높아지는 고칼륨혈증은 위약군(3.3%)에 비해 피네레논군(10.1%)에서 더 빈번하게 관찰되었으며, 이로 인해 1.7%의 환자가 치료를 중단했습니다.
일반명 피네레논으로 알려진 케렌디아는 이미 바이엘의 핵심 약물입니다. 2021년 2형 당뇨병 관련 만성 신장병 치료제로 처음 승인되었으며, 2025년에는 특정 유형의 심부전 치료제로 승인되어 아스트라제네카의 포시가(Farxiga)와 같은 약물과 경쟁하고 있습니다.
적응증 확대가 승인되면, 치료가 절실한 환자들에게 새로운 길을 열어주는 동시에 바이엘의 핵심 제약 자산에 대한 새로운 수익원을 창출하게 됩니다. 투자자들은 이제 6개월의 우선 심사 기간 내에 발표될 FDA의 최종 결정을 주시할 것입니다.
본 기사는 정보 제공 목적으로만 작성되었으며 투자 조언을 구성하지 않습니다.