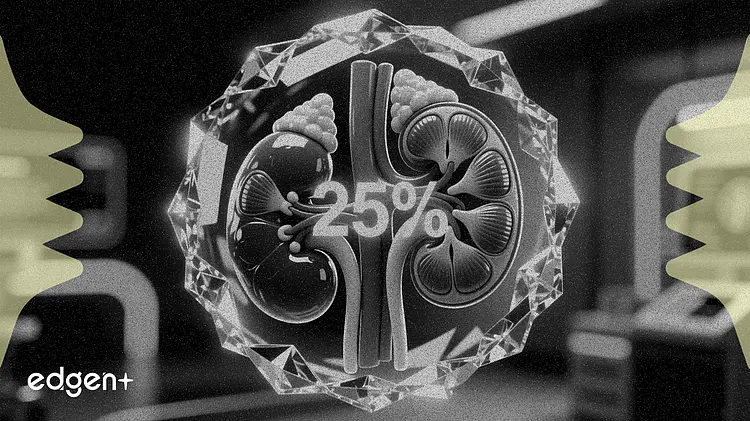Bayer AG a obtenu jeudi une désignation d'examen prioritaire de la part de la Food and Drug Administration (FDA) américaine pour son médicament Kerendia, visant à étendre son utilisation aux adultes atteints de maladie rénale chronique et de diabète de type 1 après qu'un essai a montré un bénéfice rénal de 25 %.
« L'acceptation de cette demande par la FDA souligne l'importance clinique de notre programme KERENDIA en cours, et la base de preuves croissante, à travers de larges populations de patients dans les maladies cardiovasculaires et rénales », a déclaré Carolina Aldworth, directrice médicale exécutive chez Bayer.
La demande de nouveau médicament supplémentaire (sNDA) est étayée par l'étude de phase III FINE-ONE portant sur 242 patients. L'essai a atteint son critère d'évaluation principal en montrant que le finérénone réduisait le rapport albumine/créatinine urinaire (UACR), un marqueur clé de la progression de la maladie rénale, d'une moyenne des moindres carrés de 25 % sur six mois par rapport au placebo (p < 0,001).
Un examen prioritaire raccourcit le calendrier d'évaluation de la FDA, accélérant potentiellement l'arrivée du premier antagoniste des récepteurs minéralocorticoïdes (MRA) pour les 20 à 30 % de patients diabétiques de type 1 aux États-Unis qui souffrent également de maladie rénale chronique. Cet essai réussi est le premier dans ce groupe de patients spécifique depuis les années 1990.
Le profil de sécurité dans l'essai FINE-ONE était largement conforme au profil connu du Kerendia chez les patients diabétiques de type 2. L'hyperkaliémie, ou taux élevé de potassium, a été observée plus fréquemment avec le finérénone (10,1 %) par rapport au placebo (3,3 %), entraînant un taux d'arrêt du traitement de 1,7 % pour le médicament.
Le Kerendia, connu sous le nom générique de finérénone, est déjà un médicament clé pour Bayer. Il a été approuvé pour la première fois en 2021 pour traiter la maladie rénale chronique associée au diabète de type 2, puis approuvé en 2025 pour certains types d'insuffisance cardiaque, concurrençant des médicaments comme le Farxiga d'AstraZeneca.
L'extension de l'indication, si elle est approuvée, ouvrirait une nouvelle voie de traitement indispensable et une nouvelle source de revenus pour l'un des actifs pharmaceutiques clés de Bayer. Les investisseurs attendent désormais la décision finale de la FDA, prévue dans le cadre de la fenêtre d'examen prioritaire de six mois.
Cet article est à titre informatif uniquement et ne constitue pas un conseil en investissement.